
一、 美国FDA监管信息简介
1. 监管局及简介

美国食品药品监督管理局FDA(Food and Drug Administration)是美国卫生与公众服务部直辖的联邦政府机构。其职责为药品的市场准入、上市后监管等。
2. 什么是DMF?
药品进口到美国市场必须遵循严格的法规与管控政策,包括对 OTC、处方药、原料药以及包装材料的进口管理。通常情况下,OTC 和处方药一般进行 FDA NDC 注册以及 ANDA 注册,而原料药、辅料、包材一般进行 DMF 药物主文件备案。
DMF(Drug Master File,药品文献档案)是美国FDA要求的一种非公开文件,用于提交药品、原料药、包装材料、助剂、配方和工艺等有关数据的注册文件。
DMF通常由药品原料、药品包装和助剂的制造商或供应商向FDA提交,并用于支持申请商的新药批准申请(NDA)、通用药申请(ANDA)或生物等其它申请。
3. 主要指南
DMF Guidance
FDA, 2018, Comprehensive Table of Contents Headings and Hierarchy.
FDA, 2018, eCTD Technical Conformance Guide.
FDA, 2017, MAPP 5040.1 Product Quality Microbiology Information in the Common Technical Document—Quality (CTD-Q).
FDA, 2017, Technical Conformance Guide for Shared System REMS Drug Master File Submissions.
FDA, 2017, Transmitting Electronic Submissions Using eCTD Specifications.
4. DMF的五种类型
a. Type I: Manufacturing Site, Facilities, Operating Procedures, and Personnel
I 型:生产场所、设施、操作程序和人员
b. Type II: Drug Substance, Drug Substance Intermediate, and Material Used in Their Preparation, or Drug Product
II 型:原料药、原料药中间体以及用于其制备的材料或药品制剂
c. Type III: Packaging Material
III 型:包装材料
d. Type IV: Excipient, Colorant, Flavor, Essence, or Material Used in Their Preparation
IV 型:赋形剂、着色剂、香料、香精或用于其制备的材料
e. Type V: FDA Accepted Reference Information
V 型:FDA 接受的参考信息
DMF持有人(DMF Holder)可以授权一个或者多个申请人索引同一个DMF,无需向申请人披露DMF中的机密信息;DMF的递交完全由持有人自行决定,法规不强制要求申报DMF。
5. 注册语言
英语
二、 注册流程、周期&官费
1. 注册流程图

2. 相关费用和周期
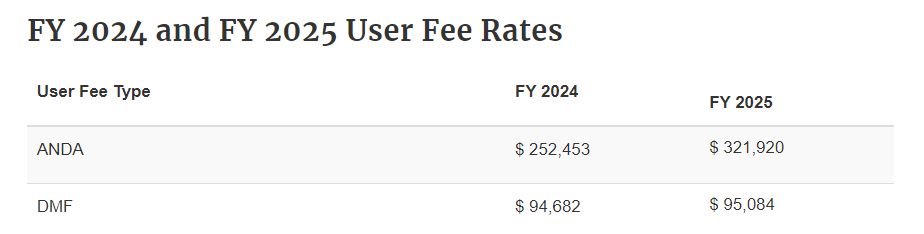
Type II 原料药DMF审评费用每个财年(10月1日至次年的9月30日)各不相同,2024财年及2025财年的DMF费用如下所示:
a. 行政审评
一般递交后2-3周内完成
b. 完整性(CA)审评
对于原料药Type I DMF需按GDUFA进行缴费; 缴费后方能进行CA审评,一般需要60天左右
三、 DMF申报资料内容简介
1. 行政信息(Module 1)
包含 DMF 持有人、代理人(如有)、制造商的信息及禁止认证。
持有人需提供名称、地址;代理人需提供详细联系方式及职责;制造商需提供名称、地址及联系人信息。同时需提交 LOA,明确授权方及授权内容,且需列出授权方列表并及时更新。
2. 质量概述(Module 2)
总结 Module 3(及适用时的 Module 4 和 5)的相关部分。
3. 生产与质量控制信息(Module 3)
按 DMF 类型组织信息,包括生产工艺、质量标准、分析方法验证、稳定性研究等内容,具体因 DMF 类型而异。
4. 非临床评估信息(Module 4)
若 DMF 包含非临床评估,如 Type IV DMF 中辅料或 Type II DMF 中杂质的非临床评估,或 Type V DMF 中的非临床评估,则需在该模块提交。
5. 临床信息(Module 5)
仅在如 Type V DMF 包含临床信息时提交。
6. 其他资料
如与 DMF 相关的标签、风险评估与缓解策略(REMS)相关文件等,根据 DMF 类型和具体情况在相应模块提供。例如,与药物物质、药物产品相关的 DMF 需在 Labeling 部分提供运输标签副本,有 REMS 相关内容时在 REMS 部分提供相关文件。
四、DMF amendments
1. DMF 的任何变更、信息增加或删减(包括授权信 LOA)都必须提交给 FDA,提交应包含封面信及更新的行政和技术信息(如适用)。
2. 名称变更、收购或所有权转让:DMF 持有人名称变更(如因更名、被收购或转让所有权)时,需在行政修正案中通知 FDA,涉及代理变更的还需提交代理任命信。所有权转让时,原持有人和新持有人需分别提交转让通知和接受通知,并签署承诺声明。
五、DMF年报
DMF 年报主要用于向 FDA 保证 DMF 的时效性,包含多方面内容,具体总结如下:
1. 提交要求与编号规则
①年报不应报告 DMF 的变化,若同时有修正和年报需分别提交,使用不同的 eCTD 序列编号。
2. 内容组成
①应包含封面信,FDA 建议使用 CDER 的 DMF 网页上的模板。封面信需指定提交类型为年报,并包含其他必要信息。
②需有 DMF 持有人签署的承诺声明,表明 DMF 是最新的且持有人将遵守其中声明。声明可包含在 eCTD 第 1.2 节或随年报提交。
③包含适当的行政信息,如 DMF 持有人、代理人(如有)、制造商等信息,类似初次提交中的行政信息要求。
④列出自上次年报(或原始提交日期)以来的修正案日期,有助于 FDA 了解 DMF 的更新历史。
⑤提供授权方列表,包括当前被授权引用 DMF 信息的各方信息,以及已撤回授权的各方及其撤回日期。
3. 重要性
①有助于确保 FDA 相信 DMF 持有人遵守承诺,保证 DMF 的时效性。若未按时提交年报,可能导致 DMF 被终止。
六、DMF关闭
1. FDA关闭
若DMF未递交年报,FDA将关闭此DMF,并通知持有人或者代理。
2. DMF持有人主动关闭
可以递交一个行政增补要求关闭DMF,同时递交一个已通知所有授权人DMF关闭的声明。
七、 FAQ
Q1:哪些产品的 DMF 需要收费?
A1:只有涵盖用于仿制药申请的 API(II 类 API DMF)制造的 DMF 需要收费。具体来说,每个拥有 II 类 API DMF(DMF 持有人)的人,如果在 2012 年 10 月 1 日或之后,在仿制药提交的任何初始授权书中引用该 DMF,则需要缴纳 DMF 费用。
Q2:什么是仿制药提交?
A2:仿制药提交一词是指 ANDA、ANDA 的修订或 ANDA 的 PAS。
Q3:DMF Holder的义务有哪些?
A3:变更药品主文件所需的通知、授权查询药品主文件的人员名单、年度更新、代理人的任命、所有权转让。

