
一、 英国MHRA监管信息简介
1. 监管局及简介
MHRA :英国药品和健康产品管理局(Medicines & Healthcare products Regulatory Agency) 负责运营英国医疗器械警戒系统,包括进行市场监督、执行法规以及与英国和全球的医疗保健和监管利益相关者合作。
虽然MHRA 不对医疗器械进行认证,但MHRA 对英国批准机构进行监督,如果MHRA 认为医疗器械不安全,MHRA 有权将其从英国市场上撤下。
UKCA:UK Conformity Assessment是英国合格评定的简称,是英国产品标志,用于在英国市场(英格兰、威尔士和苏格兰)销售的某些商品(包括医疗器械)。
2. 监管法规
医疗器械的UKCA标志要求基于原欧洲医疗器械指令相关的3个指令(EU AIMDD、EU MDD、EU IVDD)要求,并结合英国的医疗器械法规UK MDR 2002进行了修订,使其适用于英国。
现有的英国医疗器械法规全称为The Medical Devices Regulations 2002(简称UK MDR 2002),UK MDR涵盖MD和IVD。
基础法规:
《Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR 2002)》;
在过渡期(英国脱离欧盟后)结束前,以下法规使下列指令在英国法律中生效:
①有源植入式医疗器械指令Directive 90/385/EEC on active implantable medical devices (EU AIMDD)
②医疗器械指令Directive 93/42/EEC on medical devices (EU MDD)
③体外诊断医疗器械指令Directive 98/79/EC on in vitro diagnostic medical devices (EU IVDD)
3. 医疗器械和IVD产品风险等级分类

4. 注册依据
所有医疗器械,包括 IVD、定制设备和系统或程序包,在英国,设备必须符合Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR 2002) 法规要求,才能上市并在 MHRA 注册。即医疗器械在进入英国市场之前,必须获得 UKCA 或 CE 证书。该证书表明器械符合相关法规。
5. 注册语言
英语
6. 英国负责人(UK Responsible Person)
英国境外制造商需指定一位英国负责人(UK Responsible Person)即英国当地授权代表,来注册其产品。
①英国负责人必须提供书面证据,证明他们有制造商的授权作为其英国负责人。英国负责人代表非英国制造商执行与制造商义务相关的指定任务,这包括在设备进入英国市场之前,向MHRA注册制造商的设备。
②进口商和分销商无需指定英国负责人。
③英国负责人的职责在除注册要求外,英国负责人还必须:
a) 确保已经起草了符合性声明和技术文件,并且在适用的情况下,制造商已经执行了适当的符合性评估程序
b) 根据MHRA的请求,向MHRA提供证明设备符合性所需的所有信息和文件
c) 与MHRA合作采取任何预防或纠正措施,以消除或在不可能的情况下减轻设备带来的风险
d) 立即将医疗保健专业人员、患者和用户关于与他们指定的设备有关的疑似事件的投诉和报告告知制造商
7. 进口商和分销商
①如果英国进口商不是英国负责人,则进口商必须告知相关制造商或英国负责人其进口设备的意图。在这种情况下,制造商或制造商的英国负责人必须向MHRA提供设备进口商的详细信息。有关设备注册的进一步指南。
②有关CE标志或UKCA标志的存储,运输和检查设备标签的义务也适用。进口商或分销商的名称和地址无需出现在标签上。
二、 注册流程
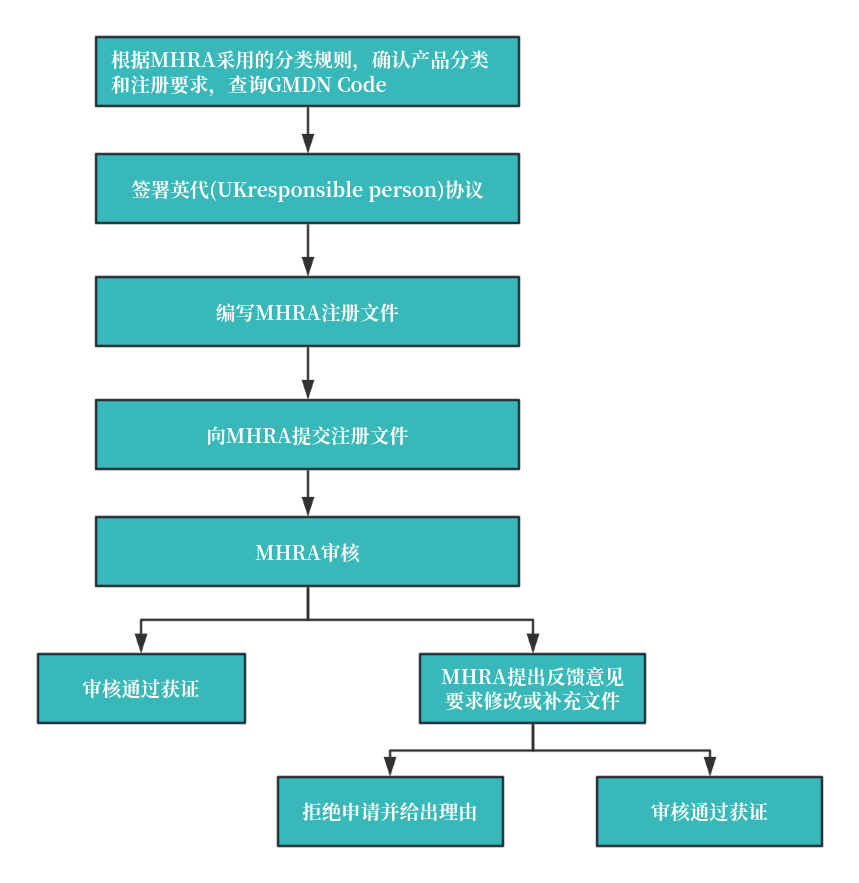
官方费用:
以下费用仅涵盖MHRA注册,对于需要由公告机构颁发证书的产品,其认证费用并未包含在内;官方费用后续可能会有所变动,具体收费标准以官方发布的最新信息为准。

三、 FAQ
Q:医疗器械产品在英国注册需要多长时间?
A:低风险的产品申请Class I & General IVDs,平均审核时间是2-4周,最长审理时间会达到90天。
如果是需要Approval Body审核的产品,需要根据具体的不同AB的审核时间来确定。
Q:医疗器械进行英国注册需要QMS证书吗?
A:医疗器械在英国注册时有需要出具单独的QMS证书。
Q:英国的官方收费是怎样的?
A:费用来源:MHRA
https://www.gov.uk/government/publications/mhra-fees/current-mhra-fees)
Q:医疗器械在完成注册后如果发现授权代表不配合业务开展,可以更换授权代表吗?
A:可以,MHRA允许制造商更换新的授权代表。
Q;一个产品可以同时被多个授权代表注册吗?
A:不可以,一个产品只允许拥有一个授权代表。
Q: 医疗器械产品完成注册后可以进行注册变更吗?
A:如果医疗器械产品变更符合MHRA允许的范围,可以由授权代表提交变更申请,经由MHRA审批后,可以投入英国市场。
Q:“批量上传”功能是否可用于所有GMDN Code组合?
A:不能,批量上传不能用于所有GMDN Code组合。“批量上传”功能只能用于某一GMDN Code的器械型号。例如, 注册1个GMDN Code,包括50种不同的器械型号,可以使用批量上传来注册这50种型号的器械。
Q:制造商是否必须在标识和包装上体现UK RP名称和地址,目前的标识和包装是否直至2023年6月30日有效?
A:目前正处于过渡阶段,如果延期法规不变,欧盟MDD / IVDD或欧盟MDR / IVDR在英国上市的器械标识和包装在2023年6月30日之前无需更改。因此目前的标识和包装(包括CE标记)仍然有效,而直到2023年6月30日,英国RP的名称和地址都强制要求体现在标识和包装上。

