
一、 沙特SFDA监管信息简介
1. 监管局及简介

沙特的监管当局是Saudi Food & Drug Authority(简称SFDA)。其主要职责是规范、监督和监测食品、药品、医疗器械、化妆品、饲料、烟草、农药,以及制定其强制性的标准规范,无论它们是进口的还是在当地生产的。
2. 监管法规
[Medical Devices Law] <医疗器械法>
[Implementing Regulation of Medical Devices Law] <医疗器械法实施条例>
3. 医疗器械和IVD产品风险等级分类
沙特根据医疗器械风险等级从低到高,将医疗器械MD、体外诊断试剂IVD分为A、B、C、D类。
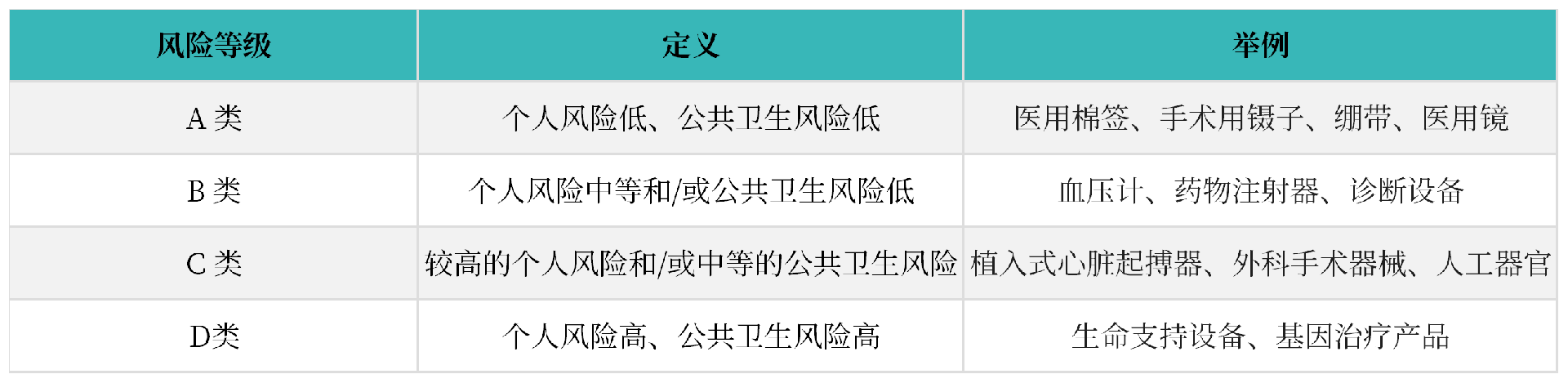
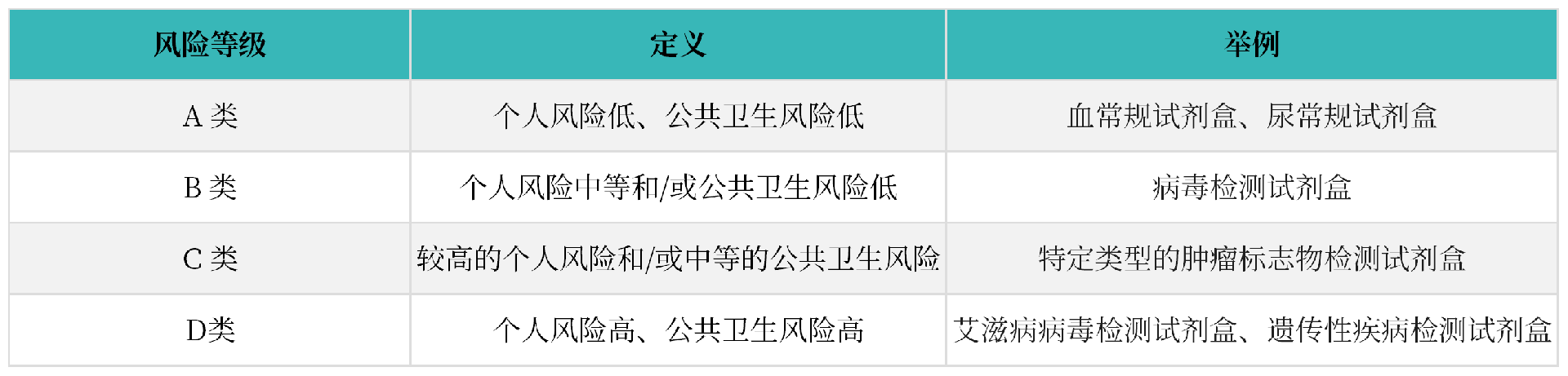
沙特根据医疗器械风险等级从低到高,将体外诊断试剂IVD分为:
4. 准入必要条件
产品清单、设计描述、配件、材料、图纸、包装、照片、风险分析等各类报告,以及证书、计划、测试报告、临床评价、检查表、声明等内容,涵盖了从产品本身信息到各类认证和评估要求的诸多方面,以确保医疗器械符合相关标准和规定,保障其安全有效上市及使用。
沙特并不强制要求CE,但是若产品标签上有CE标志,则需要提交证据支持。
5. 注册语言
英语如果医疗器械是供家庭使用或非专业人员使用,则需要提供标签、使用说明和营销材料的阿拉伯语翻译件。
6. 沙特授权代表
授权代表(Authorized representative-AR)指在沙特KSA内设立的任何自然人或法律的,并收到制造商书面授权,代表其执行特定任务,包括代表制造商与SFDA沟通的义务。
职责:
①必须在沙特境内需要有AR营业执照MDEL,若授权代表代理多个制造商,需对不同的制造商申请单独的营业执照。若MDEL有任何更新,需告知SFDA。AR营业执照MDEL有效期为1年。申请人可以在AR营业执照到期前60天内申请续证。
②需要有一份书面授权,其中规定了授权代表的指定职责。建立并遵守授权活动的相关程序SOP,需要满足质量管理体系的要求。
③制造商可以选择是指定一名授权代表代表所有医疗器械,或是为每个器械类别指定不同的授权代表。
二、 注册流程、周期&官费
1. 注册流程图
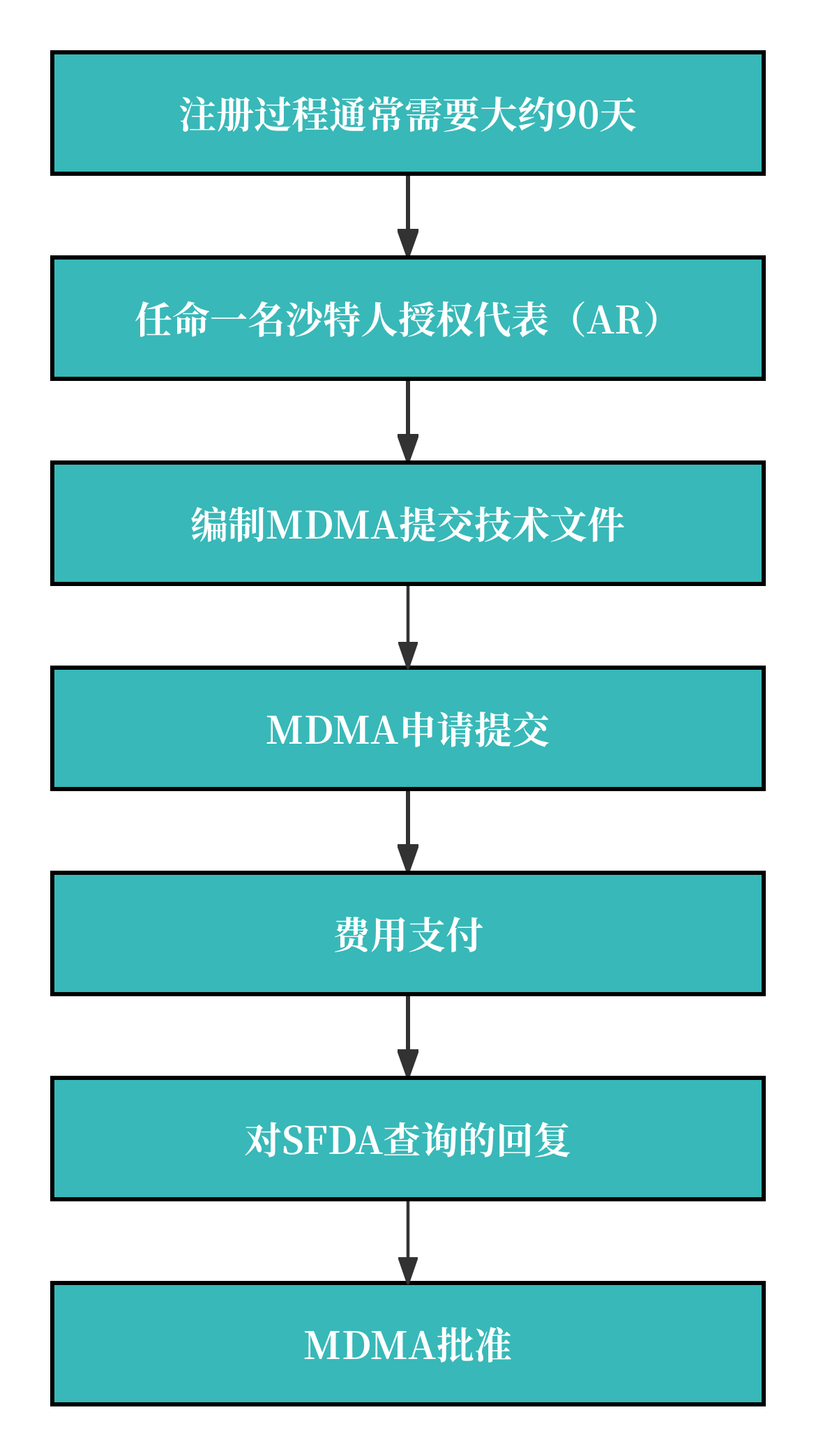
2. 注册周期及官费
注册周期:MDMA审批时间为提交申请资料后2-3个月,更高风险等级的产品审批时间会更长。
官方费用:
AR年费:1840USD
Class A 的注册评估费: SAR 15000
Class B 的注册评估费: SAR 19000
Class C 的注册评估费: SAR 21000
Class D 的注册评估费: SAR 24000
三、 FAQ
Q:什么是经营许可证(LTO)? 清关文件(符合性声明)是否与注册文件相同?
A:清关文件不同于“货物符合医疗器械和产品控制法规的声明”,后者提到了货物中产品的注册和许可信息,以及制造商确认产品是由进口商运送的文件。
Q:上市后评估研究是什么意思?
A:这些是使用科学方法对医疗器械进行的研究,以验证其安全性/有效性,因为当地销售的设备上存在安全信号。
Q:上市后评估研究的预期结果是什么?
A:纠正措施
预防措施
要求额外的临床研究,以提供医疗器械安全性和有效性的证据
向用户和医疗保健提供者传播安全通信
Q:如果制造商在没有授权代表 (AR) 的情况下直接向买家销售医疗器械,谁负责通知受影响的买家并提供纠正行动计划和关闭?
A:制造商负责在沙特阿拉伯销售的医疗器械,以及供应商和分销商。
Q:向 SFDA 指定制造商联络官或其授权代表遵循什么程序?
A:您必须遵守医疗器械上市后监督负责人公告,签署授权协议并递交给SFDA获得ARL(授权代表证书)。
ARL需要进行海牙认证。

