
一、 肯尼亚PPB监管信息简介
1. 监管局及简介

肯尼亚医疗器械注册的监管机构是药房和毒药委员会Pharmacy and Poisons Board (PPB)。该委员会主要负责医疗器械的市场准入、合规评估以及上市后监管。
2. 监管法规
[Pharmacy and Poisons Act]
3. 医疗器械和IVD产品风险等级分类
医疗器械:
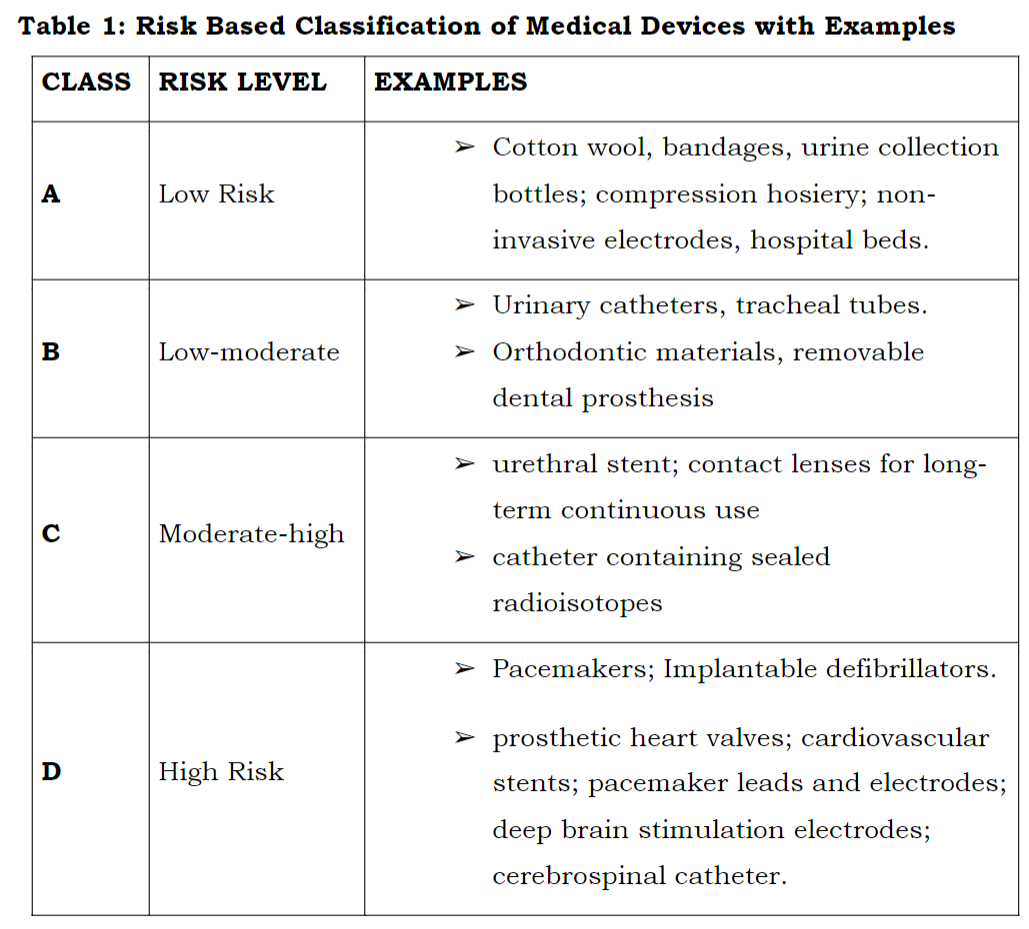
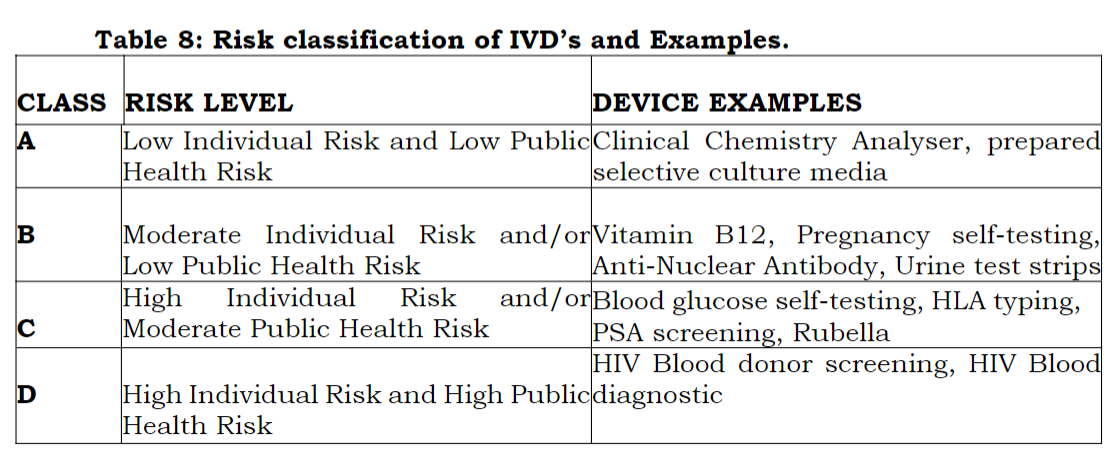
IVD:
4. 准入必要条件
ISO13485证书
委任一位肯尼亚当地授权代表(LAR)
注册文件:行政信息、分类信息、符合性声明草案、风险管理、临床评估、PMS 和 PMCF等
5. 注册语言
英语
6. 肯尼亚授权代表(LAR)
肯尼亚境外的制造商必须指定一位当地授权代表(LAR),负责与肯尼亚药房和毒药委员会(PPB)沟通。制造商需提供书面授权以证明授权代表是在其同意下行事,授权代表必须获得制造商的授权信以明确其权利。
一个制造商在肯尼亚针对同一产品线或产品组合只能指定一个授权代表。
二、 注册流程、周期&官费
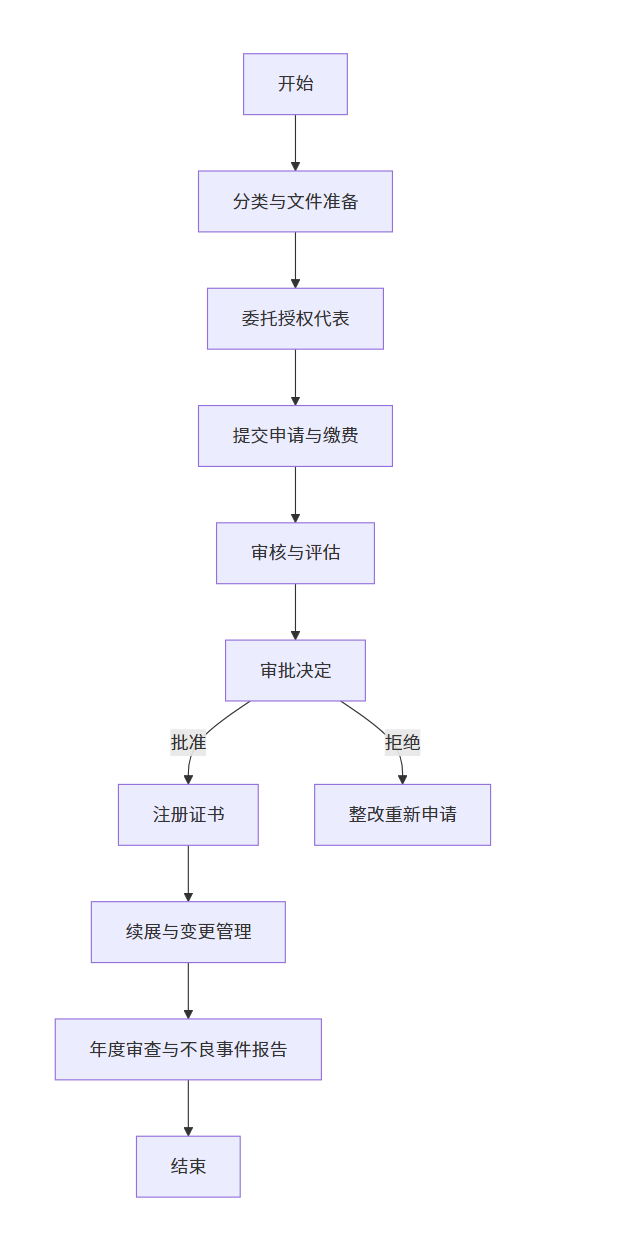
1. 注册流程图
2. 注册周期及官费
费用单位为USD

注册周期:
新注册(完整和简化评估流程)约 12 个月 (Wiselink经验)
三、 FAQ
Q:哪些医疗器械可以在肯尼亚豁免注册?
A:所有在肯尼亚销售的医疗器械都需要注册,包括含药用产品的器械、定制器械等,具体根据其风险分类确定注册要求,风险等级从 A 类到 D 类不等,不同类别适用不同的注册流程和标准。
Q:注册申请一般需要提交哪些文件?
A:需使用CSDT模板提交,包括行政信息、制造商信息、本地授权代表信息、技术文件摘要等,不同类别器械还需额外提供如授权信、标签等特定文件。
Q:医疗器械注册有哪些评估路线?
A:B类器械有全面审核、简化评估、快速注册(EBR)和即时注册(IBR);C 类和 D 类器械有全面审核、简化评估和快速评估路线。
Q:含药用产品的医疗器械如何注册?
A:若主要作用方式为药品则按药品监管。
Q:肯尼亚医疗器械注册证书的有效期是多久?
A:一般有效期为 5 年,但对于某些特殊情况,如注册有效期为 2 年的器械则按特殊规定执行,有效期届满前需按程序申请续展注册。
Q:医疗器械应如何变更?
A:根据变更类别:轻微变更只需提交变更通知,重大变更(如设计、原材料、预期用途等变更)需提交新的CSDT格式申请。

