FDA 510(k)是美国食品药品监督管理局(Food and Drug Administration,简称FDA)对医疗器械进行监管的一种上市前通知(Premarket Notification)程序。根据美国联邦食品、药品和化妆品法案(FD&C Act)第510(k)条款,医疗器械制造商在将产品推向市场之前,需要向FDA提交510(k)申请,以证明其产品与市场上已有的、被认可的类似产品(称为“对比器械”)具有“实质等同性”(Substantial Equivalence)。简单来说,就是新器械在安全性和有效性方面与已有的对比器械相当,没有显著差异。
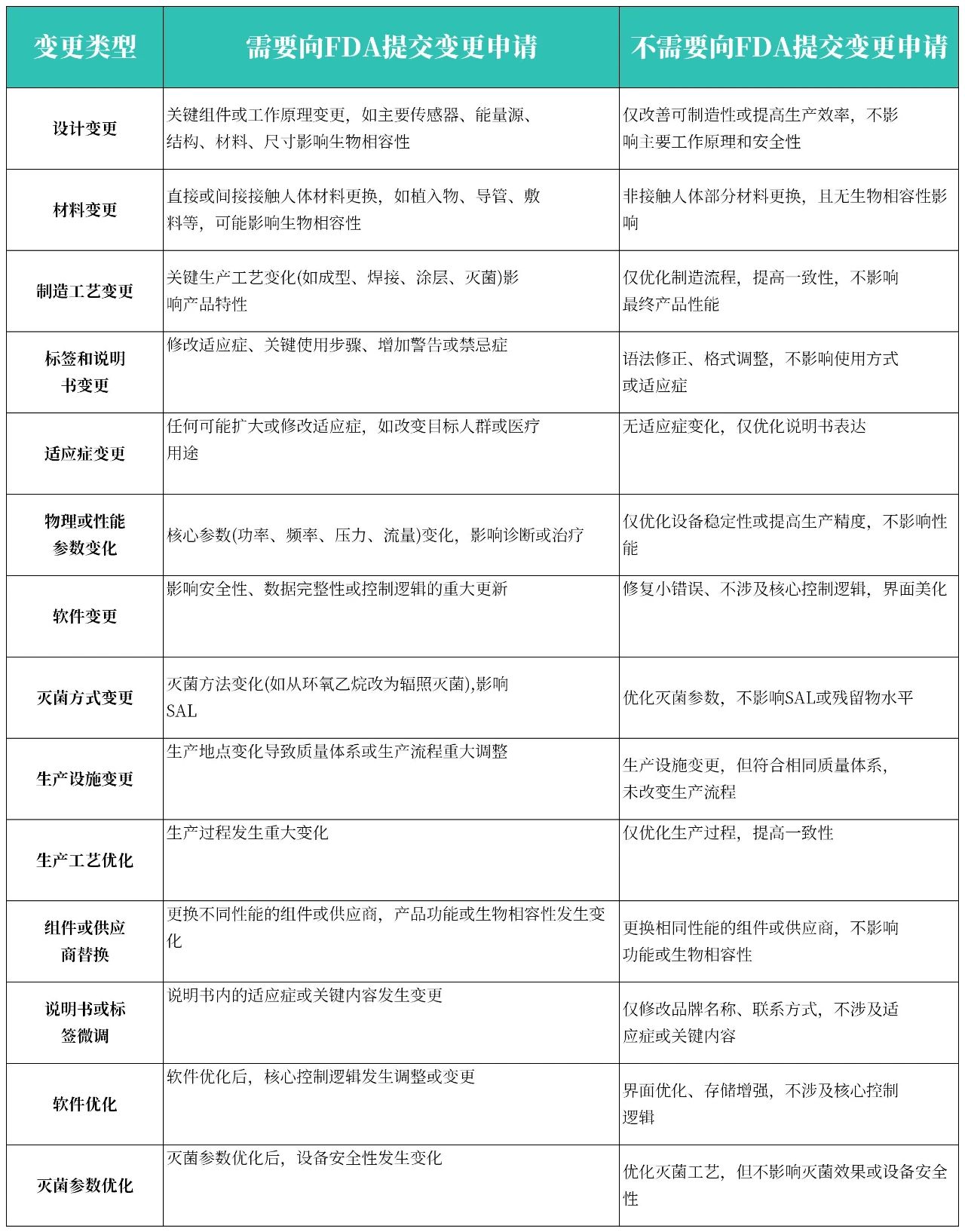
对于已经获得FDA 510(k)批准的医疗器械而言,制造商在产品变更时常常面临一个关键问题:哪些内容的变更情况需要向FDA提交变更申请,哪些内容的变更情况不需要向FDA提交变更申请呢?本篇文章,知汇用一张图给您详细介绍一下!