IVD体外诊断试剂注册是指将IVD体外诊断试剂产品提交给相关的监管机构(如FDA、CE、NMPA等),并经过审核、评估、认证后获得许可证书,允许在市场上销售和使用的过程。这个过程需要遵循相关的法律法规和规范要求,包括ISO 13485质量管理体系、ISO 14971风险管理体系、GMP(Good Manufacturing Practice)等。
IVD体外诊断试剂注册的过程包括申请、审核、评估、许可证颁发等环节。申请人需要提供产品相关的资料和证明文件,包括产品技术规格、安全性评估、临床试验数据、质量保证计划等。监管机构会对这些文件进行审核和评估,评估结果可能包括通过、需要补充材料、拒绝等。如果被批准,申请人可以获得许可证书,获得销售和使用的权利。
其分类方式可能因国家、地区、行业等不同而有所差异。根据其特点和用途,体外诊断试剂可以分为以下类型:
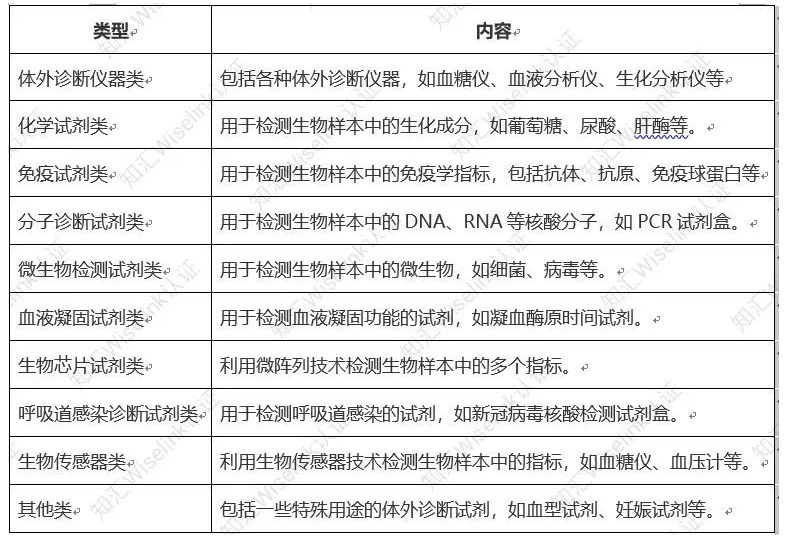
| 类型 | 内容 |
|---|---|
| 体外诊断仪器类 | 包括各种体外诊断仪器,如血糖仪、血液分析仪、生化分析仪等。 |
| 化学试剂类 | 用于检测生物样本中的生化成分,如葡萄糖、尿酸、肝酶等。 |
| 免疫试剂类 | 用于检测生物样本中的免疫学指标,包括抗体、抗原、免疫球蛋白等。 |
| 分子诊断试剂类 | 用于检测生物样本中的DNA、RNA等核酸分子,如PCR试剂盒。 |
| 微生物检测试剂类 | 用于检测生物样本中的微生物,如细菌、病毒等。 |
| 血液凝固试剂类 | 用于检测血液凝固功能的试剂,如凝血酶原时间试剂。 |
| 生物芯片试剂类 | 利用微阵列技术检测生物样本中的多个指标。 |
| 呼吸道感染诊断试剂类 | 用于检测呼吸道感染的试剂,如新冠病毒核酸检测试剂盒。 |
| 生物传感器类 | 利用生物传感器技术检测生物样本中的指标,如血糖仪、血压计等。 |
| 其他类 | 包括一些特殊用途的体外诊断试剂,如血型试剂、妊娠试剂等。 |
确定试验方案和研究设计
在设计试验方案和研究设计时,需要考虑试验的目的、研究对象、样本大小、研究方法、数据分析等方面的问题,并确保试验方案符合伦理和法律要求。
选择合适的注册平台
国际上已经建立了多个体外诊断试剂临床试验注册平台,如ClinicalTrials.gov、EU Clinical Trials Register等。选择合适的平台需要考虑平台的可靠性、注册流程和费用等方面的问题。
注册试验并提交相关信息
在注册试验时,需要提交试验方案、研究设计、研究目的、试验结果等信息,并确保信息的准确性和完整性。注册后,试验方案将被公开,并可以在注册平台上进行搜索和查看。
获得伦理委员会的批准
在进行体外诊断试剂临床试验前,需要获得伦理委员会的批准。伦理委员会需要审查试验方案和研究设计,以确保试验符合伦理要求和法律要求。
开始试验
在试验进行过程中,需要遵循试验方案和研究设计,收集、分析和报告试验数据。
更新注册信息
在试验进行过程中,如果试验方案或研究设计发生了变化,需要及时更新注册信息,并向注册机构提交更新后的信息。
完成试验并提交评价信息
完成试验后,需要对试验结果进行评价和分析,并将结果提交给相关机构进行审查和批准。这些机构可以是监管机构、学术机构、医疗机构等,其主要任务是确保试验结果的科学性和可信度,以保障公众健康和安全。
国际IVD体外诊断试剂注册需要准备的资料可能因国家和地区而异,但一般包括以下内容:
产品注册申请表:申请表需要填写产品的基本信息,包括产品名称、型号、用途、适用人群等。
产品说明书:产品说明书需要详细描述产品的性能、使用方法、注意事项等。
产品标签和包装:产品标签和包装需要符合当地的标准,包括标签上的文字、图案、条形码等。
产品检测报告:需要提供产品的检测报告,包括产品的性能指标、安全性、有效性等。
生产厂家的质量管理体系证明:生产厂家需要提供质量管理体系证明,如ISO 13485认证证书等。
生产厂家的营业执照和生产许可证:需要提供生产厂家的营业执照和生产许可证等相关证明文件。
产品销售许可证:有些国家和地区要求产品销售前需要获得当地的销售许可证,需要提供相应证明文件。
其他相关证明文件:根据当地的规定,可能需要提供其他相关证明文件,如CE注册证书、FDA注册证书等。
总之,做国际体外诊断试剂临床试验注册需要注意注册平台的选择、注册信息的准确性和完整性,以及及时更新注册信息和提交评价信息等方面的问题。同时也需要注意遵守伦理和法律要求,确保试验的科学性和可信度。







































