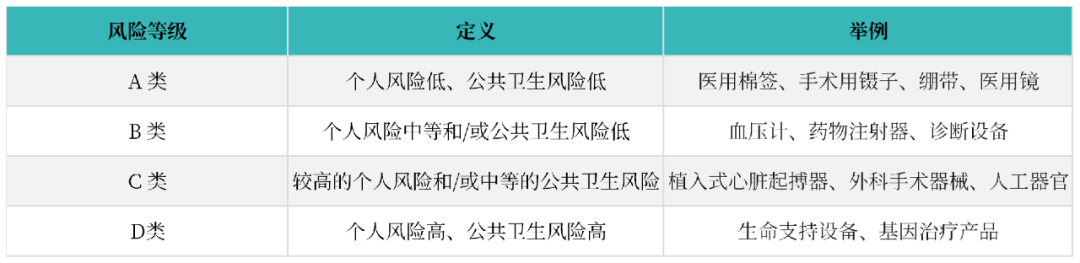
沙特食品药品管理局 Saudi Food and Drug Authority(SFDA)负责监管沙特阿拉伯的医疗器械注册。沙特医疗器械注册需要遵循 Medical Devices Law(医疗器械法),参考的法规要求是 Implementing Regulation of the Medical Devices Law(医疗器械法实施条例)。在医疗器械分类方面,沙特食品药品管理局 SFDA 将医疗器械分类为 A、B、C 或 D 级。在确定注册程序及其要求前,必须根据风险等级进行分类。申请人需要根据以下《医疗器械风险分类规则》对其设备在沙特进行分类:- 风险程度(对患者、使用者和其他人造成伤害的概率和严重程度)
相同的器械可根据目标人体部位进行不同的分类。因此,预期用途对于确定正确的分类至关重要。预期用途可以从以下文件中获取:在体外诊断试剂分类方面,沙特食品药品监督管理局也采用欧洲医疗器械法规 IVDR,将体外诊断试剂分为A、B、C、D四个等级。申请人需要根据 IVD 医疗器械分类规则,并且考虑预期用途、风险程度来确定IVD 的类别。
- 定期安全更新报告 (PSUR) – 适用于 B、C 和 D 类设备
- 符合澳大利亚、加拿大、日本、美国和欧盟其中一个或多个司法管辖区适用的相关监管要求的文件证据
- 欧盟:符合欧洲法规要求的声明 (DOC),以及在欧盟国家之一的医疗器械注册证。
- 美国:510k 许可或制造商关于该器械符合 510(k)要求的声明。
- 加拿大:符合加拿大法规要求的声明 (DOC) 和企业许可证,出口到 KSA 的医疗器械的制造商证书。
- 澳大利亚:TGA 符合性评估证书,符合澳大利亚法规要求的声明(DOC)。
- 日本:符合日本监管要求的自我声明和日本医疗器械上市通知书。
在 SFDA 注册您的设备的第一步是任命沙特阿拉伯的当地授权代表 (LAR)。LAR 是一个法人实体,拥有您(制造商)的书面授权,代表您在该国执行 SFDA 医疗器械法律和法规。LAR 协议受沙特阿拉伯法律管辖,必须记录在案、获得批准并在 SFDA 注册。LAR 负责提交医疗器械营销授权 (MDMA) 的申请和文件,以及与 SFDA 沟通并处理任何上市后问题。LAR 必须满足以下特定要求 (MDS - REQ 1,医疗器械营销授权要求):
- 提交质量管理体系应用证据或 SFDA 的检查报告,以证明其符合沙特标准(SFDA.MD/GSO ISO 13485 )或同等标准的要求;
- 确保没有为同一类或同一组医疗器械指定其他 LAR;
- 将执行分配给他们的任务所需的流程记录下来,并附上相关文件。
如果您想了解您的医疗器械产品具体分类,可以咨询Wiselink知汇。技术文档是强制性文件,必须以清晰、有条理、易于搜索且无歧义的方式呈现。制造商提供的信息应满足安全与性能基本原则的要求。低风险医疗器械(除体外诊断器械、无菌器械、具有测量功能器械、可重复使用手术器械、新型器械外)应按照《技术资料》(附件3)要求提交以下材料:
值得注意的是,即使在获得 MDMA 后,SFDA 也有权在必要时要求提供所有“技术文件”,制造商应在 SFDA 提出要求后 10 天内提供所要求的文件。满足所有要求后,将颁发企业许可证,有效期为一年或类似的可续期期限。国家食品药品监督管理局有一个名为 MDMA 电子服务的在线系统,您可以在其中注册您的设备和制造商、提交您的 MDMA 申请、支付费用并跟踪您的申请状态。您需要在 MDMA 电子服务上注册您的设备和制造商,然后才能提交申请。要申请医疗器械的营销授权,申请人必须通过“GHAD 系统 - 营销授权服务”门户以电子方式提交“医疗器械营销授权申请表”。还必须提供申请表中指定的所有必需支持文件。获得机构注册号码后,您需要通过 GHAD 系统提交新的许可证申请,上传许可证申请所需的文件,如果一切正常,则进入付款部分。国家食品药品监督管理局将对申请进行技术审查,并可能要求提供更多信息或证据以验证设备的安全性和性能。国家食品药品监督管理局可能会要求:
一旦 SFDA 批准申请,将颁发阿拉伯语和英语的 MDMA 证书。MDMA 证书是授权医疗器械或医疗器械组在沙特阿拉伯营销的官方文件。MDMA 证书包含以下信息:
- 产品清关不需要代理商授权进口商去进口,只要有 MDMA 证书即可清关,清关时不需要 AR 证书。
- 例如产品代理商 A 是产品的 AR ,制造商或品牌商有产品的 MDMA 证书,在双方合同未表明产品代理商 A 为产品独代的前提下,制造商或品牌商可以授权其他代理商进行产品的进口。