
日本医疗器械监管由厚生劳动省MHLW(Ministry of Health,Labor and Welfare)和药品和医疗器械局PMDA协同负责,MHLW为核心监管部门,负责制定监管政策、最终审查注册批准、产品退市等核心管理工作;PMDA为相对独立的行政机构,与MHLW合作实施监管政策,主要承担医疗器械审核审批、QMS/GLP/GCP体系检查、不良事件报告收集分析等工作。
日本PMD法案官网:https://www.japaneselawtranslation.go.jp/ja/laws/view/3214
大部分有基准II类,小部分有基准III类,由经厚生劳动省批准的Registered Certification Body为其颁发认证。以下是其中较具代表性的机构。

Pharmaceutical and Medical Device Act,简称PMD act.

日本医疗器械分为I、II、III、IV 4个风险等级,按风险程度从低到高划分。

日语

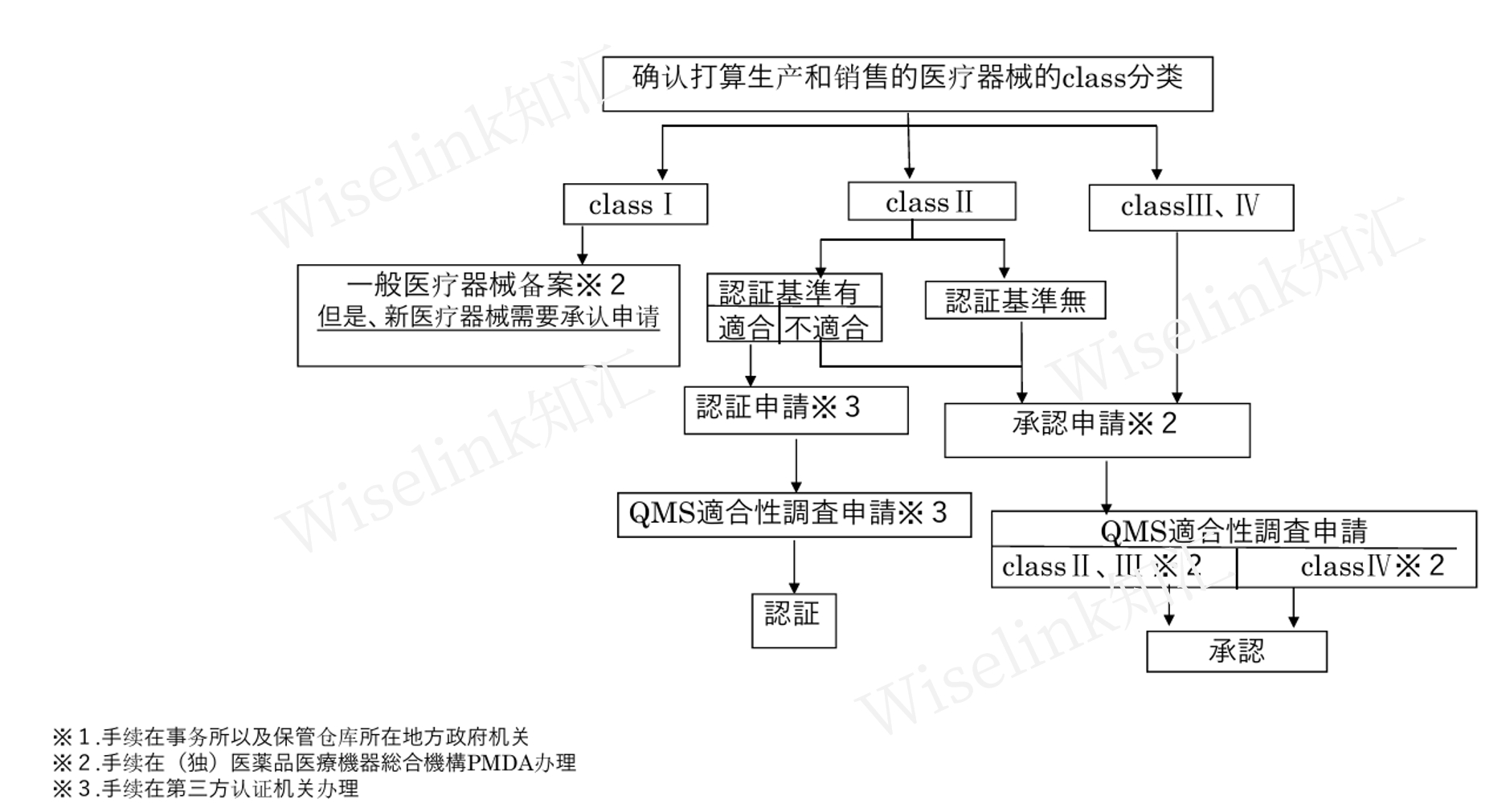
1. Class 1医疗器械备案流程
产品备案申请材料制作
向PMDA提交申请
PMDA受理盖章结束
2. Class 2/ Class 3 有基准医疗器械认证流程
(1)产品本身的审批
申请材料制作(需准备别纸1-7及各类别添资料,包括使用目的、形状原理、原材料、性能安全、使用方法、添付文件等)
申请材料提交
对认证机关所提出指示进行整改
审批结束
(2)QMS品质管理体系审核
审查资料提出(包括ISO13485认证书、年审报告、体系手册等)
书面审批(有ISO13485认证的情况)
中国厂家的QMS品质管理体系调查、进口公司的QMS品质管理体系调查(日本保管仓库也需要QMS审查)
完善QMS体系手册以及记录
书面审批或者现场审批
对认证机关所提出指示进行整改
审批结束(PMDA综合判断)
3. Class 2/ Class 3 无基准以及Class 4 医疗器械承认流程
前期沟通:PMDA各种面谈确定申请区分、是否需要临床及相关报告的可用性等;可进行竞品对比,如需临床则开展临床试验(部分环节可省略)。
产品本身的审批:申请材料制作→ 申请材料提交 → 对PMDA所提出指示进行整改 → 审批结束。
QMS品质管理体系审核:流程如上QMS品质管理体系审查
审查周期
以下为不同情况下平均完成时间的估算(提交至审查方后):
Class I- Notification备案:0 days
Class II-Certification认证:4-9 months
Class III-具有认证标准,不做临床试验:9-12 months
Class III-无认证标准,不做临床试验:12 months(me-too 仿制) or 15 months(improved 改进)
Class III-无认证标准(改进),做临床试验:16 months
ClassIII-无认证标准(新),做临床试验:16 months
外国制造商注册登记(FMR):1-2 months
医疗器械质量管理体系(QMS):About 75 days
官费
由J-RCB认证机构的费用:不同机构不同产品均会影响,单个产品可能会在100万-150万日元
由PMDA审核的费用:
PMD法案第63条规定医疗器械直接容器或包装需标注以下信息:
①营销授权持有人的姓名和地址
②产品名称
③制造编号或制造代码
④MHLW指定医疗器械的内容物重量、体积、数量等
⑤按相关条款规定的医疗器械标准事项
⑥MHLW指定医疗器械的有效期
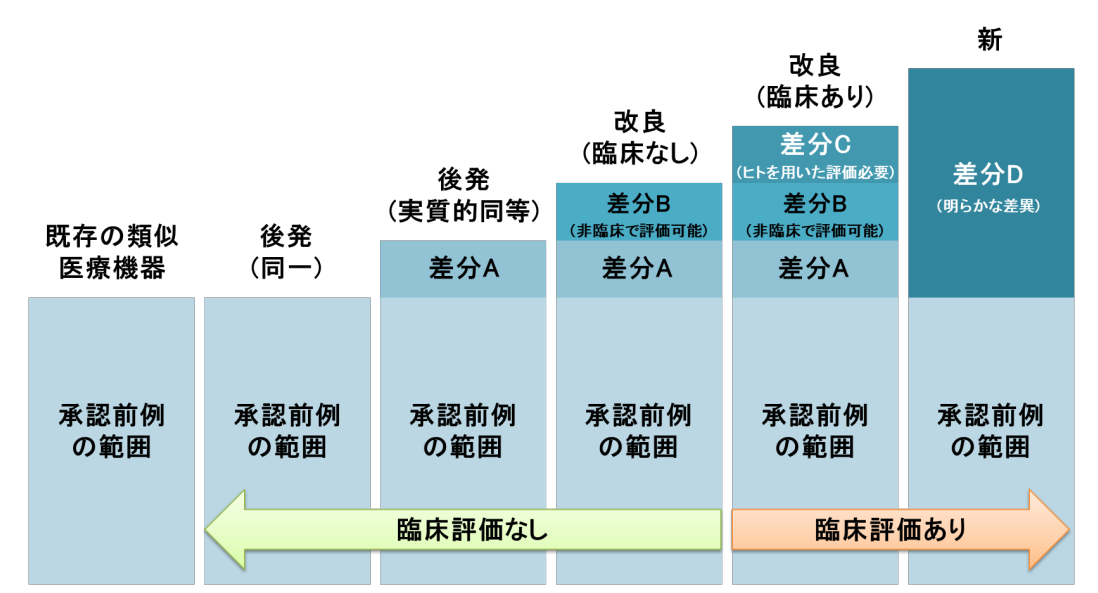
需临床:新器械、改良后有明显差异(差分C、差分D)、需用人进行临床评价的产品。
无需临床:改良后无实质差异(差分B)、后发产品(实质同等)、非临床评价可证明安全性的产品(差分A)。
核心判断依据:与既存类似医疗器械承认前例的差异程度,是否需要通过临床数据证明产品安全有效。








































